
Contente
A epilepsia é um distúrbio caracterizado pela comunicação descontrolada e desorganizada entre as células nervosas do cérebro. Em cerca de metade das pessoas diagnosticadas com epilepsia, a causa é desconhecida.Para a outra metade, a causa pode ser atribuída a um ou mais fatores específicos, como genética, lesão ou dano cerebral, mudanças estruturais no cérebro, certas condições e doenças e distúrbios do desenvolvimento.
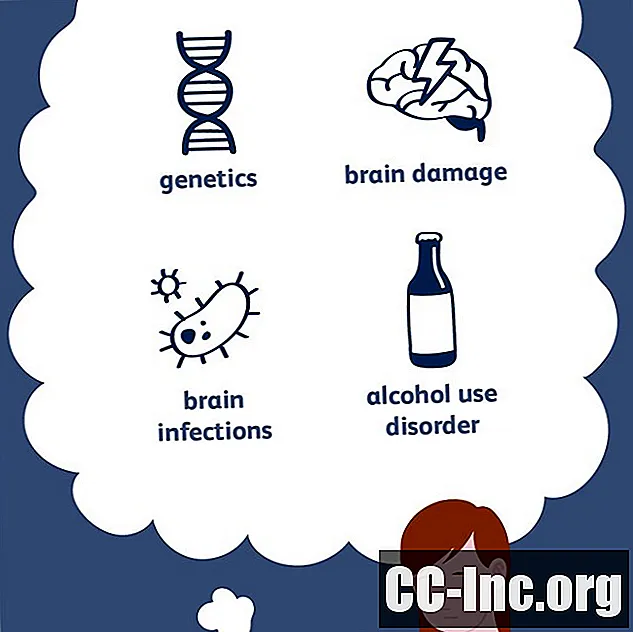
Causas comuns
A epilepsia é um distúrbio complexo com várias causas. Qualquer coisa que perturbe o padrão elétrico normal do cérebro pode levar a convulsões. Cerca de metade dos casos de epilepsia podem estar ligados a fatores específicos, incluindo:
- Genética
- Dano cerebral
- Infecções cerebrais
- Transtornos de desenvolvimento
- Mudanças estruturais no cérebro
- Álcool
Genética
A maioria das epilepsias genéticas começa na infância e é causada por um defeito genético nos canais iônicos ou receptores.
É importante observar que, para a maioria das pessoas com uma forma genética de epilepsia, os genes não são a única causa. (A genética é abordada com mais detalhes abaixo.)
Dano cerebral
Condições que causam danos ao cérebro podem causar epilepsia. Esses incluem:
- Derrame
- Tumores
- Lesões traumáticas na cabeça
- Danos cerebrais que ocorrem antes do nascimento (como por falta de oxigênio ou infecção materna)
O AVC é a principal causa de epilepsia em adultos diagnosticados após os 65 anos.
Uma Visão Geral do AVCInfecções cerebrais
Alguns casos de epilepsia são causados por infecções que afetam e inflamam seu cérebro, como:
- Meningite
- Encefalite viral
- Tuberculose
- Síndrome de imunodeficiência adquirida (AIDS)
Distúrbios do Desenvolvimento
A epilepsia parece ser mais comum em pessoas com certos distúrbios de desenvolvimento, incluindo:
- Autismo
- Síndrome de Down
- Paralisia cerebral
- Deficiência intelectual
Mudanças estruturais no cérebro
Certas diferenças na estrutura do cérebro podem causar convulsões, incluindo:
- Esclerose hipocampal (um hipocampo encolhido, uma parte do cérebro que desempenha um papel importante na aprendizagem, memória e emoções)
- Displasia cortical focal (anormalidade do desenvolvimento do cérebro em que os neurônios não conseguem migrar para o local apropriado)
Álcool
Alguns estudos mostraram que o abuso crônico de álcool pode estar associado ao desenvolvimento de epilepsia em algumas pessoas. Essa pesquisa sugere que as crises repetidas de abstinência do álcool podem tornar o cérebro mais excitável com o passar do tempo. Além disso, essa população também apresenta maior incidência de lesão cerebral traumática, que também pode causar epilepsia.
Genética
Se a epilepsia é comum em sua família, provavelmente é devido a um componente genético. Algumas epilepsias com causas desconhecidas também podem ter um componente genético que ainda não é conhecido.
Embora alguns genes específicos estejam ligados a certos tipos de epilepsia, na maioria dos casos, os genes não necessariamente causa epilepsia - eles podem apenas aumentar a probabilidade de ocorrer nas circunstâncias certas.
Se você sofrer um traumatismo cranioencefálico e tiver histórico familiar de epilepsia, por exemplo, é mais provável que você a desenvolva. Os genes são apenas uma peça do complexo quebra-cabeça para a maioria das pessoas.
Vários tipos e síndromes de epilepsia específicos são conhecidos por terem um componente genético.
Epilepsia Neonatal Familiar
As convulsões geralmente começam entre quatro e sete dias após o nascimento do bebê e a maioria pára por volta de seis semanas após o nascimento, embora possam não parar até os 4 meses de idade. Alguns bebês também podem ter convulsões mais tarde na vida.
Mutações no gene KCNQ2 são mais frequentemente a causa, embora mutações no gene KCNQ3 também possam ser um fator.
Epilepsia genética com convulsões febris Plus (GEFS +)
GEFS + é um espectro de distúrbios convulsivos. As convulsões geralmente começam entre as idades de 6 meses e 6 anos, quando a criança tem febre, chamada de convulsão febril.
Algumas crianças também desenvolvem convulsões sem febre, geralmente convulsões generalizadas, como de ausência, tônico-clônicas, mioclônicas ou atônicas. As convulsões geralmente param no início da adolescência.
SCN1A, SCN1B, GABRG2 e PCDH19 são alguns dos genes que foram associados a GEFS +.
Síndrome de Dravet
Esta síndrome é considerada o lado grave do espectro GEFS +. As convulsões geralmente começam por volta dos 6 meses de idade. Muitas crianças com essa síndrome têm sua primeira convulsão quando têm febre.
Também se desenvolvem convulsões mioclônicas, tônico-clônicas e de ausência atípica, que são difíceis de controlar e podem piorar à medida que a criança cresce. A deficiência intelectual é comum.
Mais de 80% das pessoas com síndrome de Dravet apresentam mutações no gene do canal de sódio SCN1A.
Síndrome de Ohtahara
Nessa síndrome rara, as crises tônicas geralmente começam no primeiro mês após o nascimento, embora isso possa acontecer até três meses depois.
Um em cada três bebês também pode desenvolver convulsões focais, atônicas, mioclônicas ou tônico-clônicas. Embora raro, esse tipo de epilepsia pode ser fatal antes dos 2 anos de idade. Algumas crianças podem desenvolver posteriormente a síndrome de West ou a síndrome de Lennox-Gastaut.
Vários genes foram associados à síndrome de Ohtahara, incluindo STXBP1, SLC25A22, CDKL5, ARX, SPTAN1, PCDH19, KCNQ2 e SCN2A.
Epilepsia Mioclônica Juvenil
Uma das epilepsias generalizadas mais comuns com um componente genético, a epilepsia mioclônica juvenil consiste em crises tônico-clônicas, de ausência e mioclônicas que começam na infância ou adolescência, geralmente entre as idades de 12 a 18 anos. As crises tendem a ser saudáveis -controlado com medicação e parece melhorar quando chega aos 40 anos.
Os genes associados a essa síndrome são CACNB4, GABRA1, GABRD e EFHC1, embora os padrões tendam a ser complexos.
Epilepsia do lobo frontal noturna autossômica dominante
As convulsões geralmente começam por volta dos 9 anos de idade e a maioria começa aos 20 anos. Elas ocorrem rapidamente, várias vezes durante o sono, e variam desde simplesmente acordar você até causar gritos, errantes, contorções, choro ou outras respostas focais.
Embora essa síndrome seja vitalícia, as crises não vão piorar e podem se tornar menos frequentes e mais leves com a idade. Eles também costumam ser bem controlados com medicamentos. Essa epilepsia não é muito comum e quase sempre é hereditária.
Mutações nos genes da subunidade do receptor nicotínico CHRNA4, CHRNB2, CHRNA2 e DEPDC5 foram associadas a essa síndrome.
Epilepsia de ausência infantil
As crises de ausência geralmente começam entre as idades de 2 e 12 anos e geralmente são genéticas. Em cerca de 2 em cada 3 crianças, as convulsões param na adolescência. Alguns desenvolvem outros tipos de convulsões.
Os genes associados à epilepsia de ausência na infância incluem GABRG2 e CACNA1A.
Epilepsia de ausência juvenil
Esta síndrome começa mais tarde na vida e as crises tendem a durar mais tempo do que na epilepsia de ausência da infância. Geralmente também é uma condição para a vida toda, enquanto as crianças com epilepsia de ausência na infância tendem a superar os ataques.
As crises de ausência geralmente começam entre as idades de 9 e 13 anos, embora possam começar em qualquer idade entre 8 e 20 anos. As crises tônico-clônicas, normalmente ao acordar, também são vistas em cerca de 80% das pessoas com essa síndrome.
A causa costuma ser genética, e os genes ligados à epilepsia de ausência juvenil são GABRG2 e CACNA1A, entre outros.
Epilepsia com crises generalizadas tônico-clônicas isoladas
As convulsões tônico-clônicas podem começar em qualquer lugar entre 5 e 40 anos de idade, embora a maioria comece entre 11 e 23 anos. As convulsões geralmente acontecem duas horas após o despertar.
A privação do sono, fadiga, álcool, menstruação, luzes piscando e febre costumam ser os gatilhos, e a maioria das pessoas precisará de medicamentos para o resto da vida.
O principal gene associado a essa síndrome é o CLCN2.
Epilepsia familiar do lobo temporal
Se você tem crises focais que começam no lobo temporal e uma história familiar de crises semelhantes, é considerado que você tem essa síndrome. As convulsões tendem a ser bastante raras e leves; tão brandos, na verdade, que podem não ser reconhecidos.
As convulsões geralmente começam após os 10 anos e são facilmente controladas com medicamentos.
O gene associado a essa epilepsia hereditária é o DEPDC5.
Epilepsia Focal Familiar com Focos Variáveis
Esta epilepsia hereditária geralmente consiste em um tipo específico de crise focal. Todos os membros de uma família com epilepsia têm um único tipo de crise focal, mas as crises podem começar em diferentes partes do cérebro.
As convulsões são geralmente fáceis de controlar com medicação e geralmente são raras.
O gene DEPDC5 também está ligado a essa síndrome.
Síndrome de West
Os espasmos infantis começam no primeiro ano de vida e geralmente param entre as idades de 2 e 4 anos.
Anormalidades nos genes ARX, CDKL5, SPTAN1 e STXBP1 foram encontradas nesta síndrome, embora outras causas incluam anormalidades estruturais do cérebro, às vezes de natureza genética, e anormalidades cromossômicas.
Epilepsia Rolândica Benigna
Também conhecida como epilepsia infantil com pontas centrotemporais, essa síndrome afeta cerca de 15% das crianças com epilepsia e é mais comum em crianças com parentes próximos que têm epilepsia. A maioria supera-a aos 15 anos.
O gene associado a essa síndrome é GRIN2A, embora este seja outro caso em que o padrão genético é extremamente complexo.
Fatores de risco
Os fatores de risco mais comuns para epilepsia incluem:
- Era: Embora possa começar em qualquer idade, a epilepsia tende a aparecer com mais frequência em crianças e adultos mais velhos.
- História de família: Se alguém em sua família tem epilepsia, o risco de desenvolvê-la pode ser maior.
- História de lesões na cabeça: As convulsões podem ocorrer horas, dias, meses ou até anos após o traumatismo craniano, e o risco pode ser maior se você também tiver um histórico familiar de epilepsia.
- Convulsões na infância: Se você teve uma convulsão prolongada ou outra condição neurológica na infância, o risco de epilepsia é maior. Isso não inclui convulsões febris, que ocorrem quando você tem febre alta, a menos que suas convulsões febris tenham sido anormalmente longas.
- Fatores de nascimento: Se você fosse pequeno ao nascer; você foi privado de oxigênio em qualquer momento antes, durante ou depois de seu nascimento; você teve convulsões no primeiro mês após o nascimento; ou você nasceu com anormalidades no cérebro, o risco de epilepsia é maior.
Gatilhos de convulsão
Certas circunstâncias ou situações podem aumentar a probabilidade de você ter uma convulsão. Eles são conhecidos como gatilhos e se você for capaz de descobrir quais são os seus, essas informações podem ajudá-lo a gerenciar e potencialmente prevenir mais ataques.
Os fatores que podem contribuir para as convulsões incluem:
- Privação de sono, seja interrompida ou perdida
- Faltando ou pulando sua medicação
- Estar doente, com ou sem febre
- Sentindo estressado
- Quaisquer medicamentos, sejam de venda livre, com receita ou suplementos nutricionais, que possam interferir na eficácia de sua medicação para convulsões
- Não recebendo vitaminas e minerais suficientes
- Baixo teor de açúcar no sangue
- Ciclos menstruais e / ou alterações hormonais, como puberdade e menopausa
- Luzes piscando ou padrões visuais específicos, como em videogames (epilepsia fotoconvulsiva)
- Certos alimentos, atividades ou ruídos
- Uso pesado de álcool ou abstinência de álcool
- Uso de drogas recreativas
Uma palavra de Verywell
A maioria dos fatores de risco para epilepsia está fora de seu controle ou é difícil de controlar. Se você acredita que está sob alto risco, limite a quantidade de álcool que ingere e seja especialmente cuidadoso quando se trata de ferimentos na cabeça.
Não importa a causa da epilepsia, você tem muitas opções para controlar suas convulsões, o que pode ajudá-lo a viver uma vida plena.
Como a epilepsia é diagnosticada