
Contente
A fibrose cística (FC) é uma doença hereditária com risco de vida que danifica os pulmões e o trato digestivo. É causada por um gene defeituoso que desencadeia a produção de muco espesso que obstrui as vias respiratórias e bloqueia a secreção de enzimas digestivas.Os sintomas são progressivos e frequentemente graves e podem incluir problemas respiratórios, infecções pulmonares recorrentes, crescimento deficiente, infertilidade masculina e inflamação crônica do pâncreas, fígado, rins e coração.
A FC pode ser diagnosticada com exames de sangue, triagem genética e um procedimento conhecido como teste de cloro no suor.
Embora não haja cura para a FC, existem tratamentos que podem melhorar a duração e a qualidade de vida de uma pessoa.
Isso inclui técnicas de desobstrução das vias aéreas, antibióticos inalados, diluentes de muco, enzimas pancreáticas, dieta hipercalórica e medicamentos de nova geração conhecidos como moduladores CFTR. Em casos graves, pode ser necessário um transplante de pulmão.
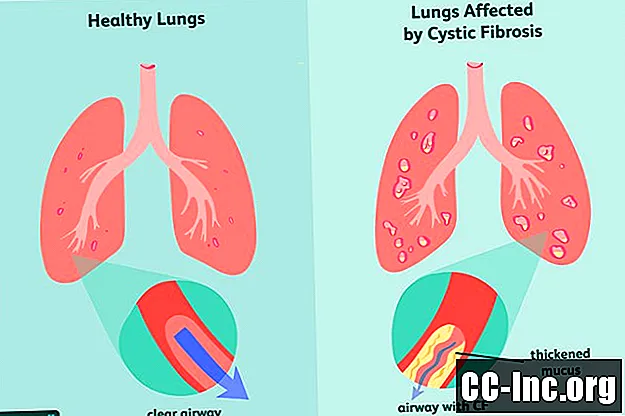
Sintomas de fibrose cística
Como um distúrbio genético, a fibrose cística é algo com o qual você nasce. Pode ou não se apresentar com sintomas no momento do nascimento e muitas vezes pode levar meses ou até anos antes que qualquer sinal de doença apareça. Nesse momento, os pulmões e o trato digestivo podem já ter sofrido danos que não podem ser desfeitos.
Os primeiros sinais e sintomas mais comuns de FC incluem:
- Bloqueio das primeiras fezes do bebê (mecônio)
- Pele com gosto salgado
- Uma tosse crônica, chiado no peito ou expectoração colorida
- Fezes soltas, gordurosas e geralmente com mau cheiro
- Infecção pulmonar, muitas vezes recorrente
- Fraco crescimento e falta de crescimento
A menos que esses sintomas possam ser controlados, o estresse nos pulmões (e a incapacidade de ganhar peso) pode ter um efeito cumulativo, afetando vários órgãos e aumentando o risco de complicações da doença.
Algumas das complicações mais características incluem:
- Puberdade retardada
- Bronquiectasia (o espessamento crônico das paredes pulmonares)
- Perda de peso
- Pancreatite (inflamação do pâncreas)
- Infertilidade masculina
- Hipertensão pulmonar (pressão alta no pulmão)
- Cálculos biliares
- Diabetes relacionada à fibrose cística
- Cor pulmonale (insuficiência cardíaca direita)
- Cirrose (cicatriz funcional do fígado)
Como a FC causa lesão progressiva em células e tecidos, qualquer dano causado aos pulmões e outros órgãos será irreversível. A morte geralmente resulta de insuficiência respiratória, seguida por insuficiência cardíaca e hepática.
Sintomas de fibrose cística
Causas
A fibrose cística é causada pela mutação do gene do receptor transmembrana da fibrose cística (CFTR), responsável pela produção da proteína CFTR. Esta é a proteína de que o corpo precisa para regular o fluxo de sal e água para dentro e para fora das células . Se a proteína estiver deformada ou com defeito, pode causar desidratação na superfície da célula, levando ao espessamento do muco circundante.
A FC é uma doença autossômica recessiva, o que significa que você precisa herdar a mutação CFTR de sua mãe e de seu pai para ter a doença. Se você herdar apenas um gene defeituoso, não terá FC, mas será portador do gene mutado.
Você pode herdar a doença se cada um de seus pais tiver uma mutação CFTR ou a própria FC. Se ambos os pais forem portadores, você terá:
- 25 por cento de chance de ter CF
- 50 por cento de chance de ser um portador
- 25 por cento de chance de não ser afetado
Por outro lado, se um de seus pais for portador e o outro tiver CF, você tem 50/50 de chance de ter CF ou ser portador.
A fibrose cística é uma das doenças genéticas mais comuns, afetando cerca de um em cada 2.500 bebês nascidos nos Estados Unidos.
É mais comum entre caucasianos e hispânicos e ocorre com menos frequência em pessoas de ascendência africana ou asiática.
Fatores de risco de fibrose císticaDiagnóstico
Existem alguns testes usados para diagnosticar a fibrose cística. Eles atuam detectando diretamente a mutação CFTR ou medindo indiretamente as alterações biológicas consistentes com a doença. O método de diagnóstico pode variar durante a gravidez, quando o bebê nasce ou a qualquer momento depois.
Guia de discussão do médico de fibrose cística
Obtenha nosso guia para impressão na sua próxima consulta médica para ajudá-lo a fazer as perguntas certas.

Dos dois testes padrão comumente usados para diagnosticar FC:
- Teste de cloreto de suor, também conhecido simplesmente como teste do suor, mede a quantidade de cloreto na pele. Como o FC interfere na transferência de sal de e para as células, haverá um acúmulo de sal no suor.
- Teste genético de CFTR é usado para detectar as mutações mais comuns da mutação CFTR. Embora existam mais de 2.000 mutações CFTR conhecidas por causar fibrose cística, as 23 incluídas no painel padrão representam os suspeitos mais prováveis.
Durante a gravidez, o teste genético CFTR pode ser usado para testar fluidos obtidos por meio de uma amniocentese ou células extraídas por meio de biópsia de vilo corial (CVS).
Triagem neonatal também é usado de forma padrão para diagnosticar FC e hoje é obrigatório em todos os 50 estados e no Distrito de Columbia. O que isso implica será diferente dependendo de onde você mora nos Estados Unidos. Se os resultados da triagem neonatal forem positivos, um teste de suor seria usado para confirmar o diagnóstico.
Como a fibrose cística é diagnosticadaTratamento
Embora não haja cura para a fibrose cística, os avanços no tratamento aumentaram a expectativa de vida das pessoas que vivem com a doença.
O objetivo do tratamento da FC é quatro vezes: prevenir infecções, manter a função pulmonar, normalizar a digestão e retardar a progressão da doença.
Entre as ferramentas terapêuticas usadas para gerenciar a FC:
- Técnicas de desobstrução das vias aéreas (ACTs) são realizados para desalojar e expelir o muco acumulado dos pulmões. As técnicas incluem tosse huff, percussão torácica ou oscilação da parede torácica.
- Uma dieta rica em gordura e calorias é usado para compensar a má absorção de gorduras, proteínas e nutrientes no intestino.
- Suplementos de enzimas pancreáticas são usados para reforçar as enzimas digestivas que o pâncreas não consegue produzir devido ao acúmulo excessivo de muco.
- Antibióticos são tomados diariamente para prevenir infecções pulmonares bacterianas.
- Mucolíticos- medicamentos usados para diluir o muco antes dos ACTs - podem ser usados.
- Moduladores CFTR são uma nova classe de medicamentos que podem corrigir certos defeitos na proteína CFTR e restaurar sua função reguladora.
- Terapia de oxigênio pode ser usado durante episódios agudos quando sua respiração estiver gravemente prejudicada.
- Nutrição enteral, também conhecida como alimentação por sonda, pode ser usada se você não conseguir sustentar o peso com uma alimentação normal.
- Transplante de pulmão é considerado quando seus pulmões não podem mais suportar a sobrevivência sem ventilação mecânica.
Lidar
Em 1938, quando a fibrose cística foi classificada pela primeira vez como uma doença, as crianças raramente viviam além do primeiro ano de vida. Na década de 1980, era possível esperar viver até 20 a 25 anos. Hoje, o quadro mudou totalmente, com pessoas que vivem bem em seus 40 e até 50 anos, se o tratamento for iniciado cedo e seguido.
Isso não quer dizer que a FC seja menos séria do que nunca. É um evento de mudança de vida, exigindo diligência e consistência não apenas para lidar com a doença, mas também viver o mais alto padrão de vida possível.
Para isso, você precisa normalizar a FC em sua vida, estabelecendo rotinas e práticas para evitar os altos e baixos que podem causar estresse e aumentar a incapacidade. Entre as considerações, você precisaria:
- Gerenciar sua nutrição. Pessoas com FC geralmente precisam do dobro de calorias diárias que outras pessoas.
- Exercite regularmente. As rotinas de condicionamento físico devem, idealmente, envolver um mínimo de 20 a 30 minutos de atividade aeróbica três vezes por semana. Encontre algo agradável que você possa fazer por toda a vida.
- Mantenha-se bem hidratado. Isso mantém os pulmões e os intestinos funcionando corretamente. Dependendo da sua idade, você deve beber pelo menos seis a oito copos altos de água por dia.
- Realize a desobstrução das vias aéreas corretamente. À medida que suas necessidades de saúde mudam, também podem mudar os tipos de ferramentas de limpeza de que você precisa. Fale com o seu pneumologista ou fisioterapeuta se você não estiver obtendo os resultados que deveria.
- Procure suporte. Além de amigos e familiares, você pode entrar em contato com o capítulo mais próximo da Cystic Fibrosis Foundation (CFF) para se conectar a uma rede de apoio em sua área.
- Procure ajuda financeira. O CFF oferece serviços que ajudam as famílias a lidar melhor com o alto custo do tratamento da FC.
Uma palavra de Verywell
Embora os exames de recém-nascidos tenham aumentado drasticamente a taxa de diagnósticos de FC em bebês, mais de 25% dos diagnósticos são feitos apenas durante a infância, adolescência e início da idade adulta.
Isso é problemático porque o diagnóstico e o tratamento precoces podem evitar muitas das complicações mais graves da FC antes que qualquer dano grave possa ser causado. Embora o tratamento não consiga interromper ou reverter a doença, pode garantir muitos mais anos livres da doença.
Para tal, é importante conhecer os primeiros sintomas da FC e falar com o seu médico se suspeitar que o seu filho pode ter a doença. Isso é especialmente verdadeiro em estados que fazem a triagem apenas com exames de sangue IRT, o que pode resultar em até 5 por cento das crianças tendo um diagnóstico atrasado ou um resultado falso-negativo, de acordo com pesquisa da Escola de Medicina e Saúde Pública da Universidade de Wisconsin .
Quais sintomas você pode esperar com a fibrose cística?